BactiSeq: Introduction
Bactiseq is a bacterial isolate whole genome sequence analysis pipeline. Bactiseq delivers analysis from Read quality analysis all the way through to gene comparision between samples. BactiSeq accepts reads from PacBio HiFi in the form of Fastq and BAM files, Nanopore reads, and Illumina reads. BactiSeq is capabale of Hybrid assembly or Long read/Short read only assembly.
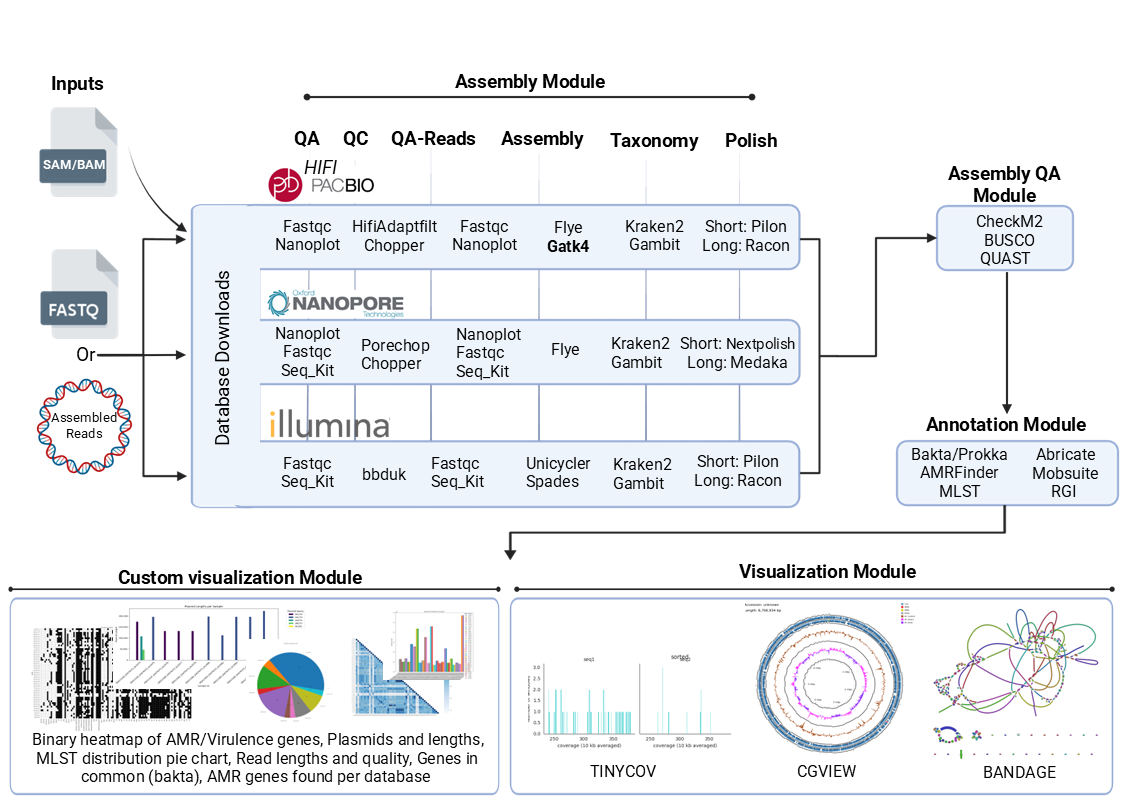
Tools used depend on the input data
Pipeline overview
The pipeline is built using Nextflow and processes data using the following steps:
Bactiseq Pipeline Workflow: Processes and Modules per Entry Data Type
| Input Data Type | Module | Software Tool(s) | Version |
|---|---|---|---|
| Pacbio HiFi | Assembly Module | ||
| if SAM/BAM file Convert to Fastq | GATK4 SamToFastq | 4.6.2.0 | |
| 1. Quality Assessment (Raw) | FastQC, Nanoplot, Seqkit stats | 0.12.1, 1.46.1, 2.9.0 | |
| 2. Quality Control & Trimming | HiFiAdaptFilt, Chopper | 3.0.0, 0.9.0 | |
| 3. Quality Assessment (Trimmed) | FastQC, Nanoplot, Seqkit stats | 0.12.1, 1.46.1, 2.9.0 | |
| 4. Assembly | Flye | 2.9.5 | |
| 5. Taxonomy Identification | Gambit, Kraken2 | 1.1.0, 2.1.5 | |
| 6. Polishing (Optional) | Pilon, Racon | 1.24, 1.5.0 | |
| Nanopore reads | Assembly Module | ||
| 1. Quality Assessment (Raw) | FastQC, Nanoplot, Seqkit stats | 0.12.1, 1.46.1, 2.9.0 | |
| 2. Quality Control & Trimming | HiFiAdaptFilt, Chopper | 3.0.0, 0.9.0 | |
| 3. Quality Assessment (Trimmed) | FastQC, Nanoplot, Seqkit stats | 0.12.1, 1.46.1, 2.9.0 | |
| 4. Assembly | Flye | 2.9.5 | |
| 5. Taxonomy Identification | Gambit, Kraken2 | 1.1.0, 2.1.5 | |
| 6. Polishing (Optional) | Nextpolish, Medaka | 1.4.1, 1.8.0 | |
| Illumina reads | Assembly Module | ||
| 1. Quality Assessment (Raw) | FastQC, Seqkit stats | 0.12.1, 2.9.0 | |
| 2. Quality Control & Trimming | bbduk | 39.18 | |
| 3. Quality Assessment (Trimmed) | FastQC, Seqkit stats | 0.12.1, 2.9.0 | |
| 4. Assembly | Unicycler, Spades | 0.5.1, 4.1.0 | |
| 5. Taxonomy Identification | Gambit, Kraken2 | 1.1.0, 2.1.5 | |
| 6. Polishing (Optional) | Pilon, Racon | 1.24, 1.5.0 | |
| Assembled Genome | Conversion Module | ||
| Convert file formats to FASTA | Any2fasta | 0.4.2 | |
| All Data | Assembly Quality Assessment Module | ||
| Quality assessment | CheckM2, BUSCO, QUAST | 1.1.0, 5.8.3, 5.3.0 | |
| Annotation Module | |||
| 1. Genome annotation | Bakta, Prokka | 1.10.4, 1.14.6 | |
| 2. AMR gene detection | RGI, AMRFinderPlus | 6.0.3, 3.12.8 | |
| 3. Virulence gene detection | Abricate | 1.0.1 | |
| 4. Plasmid detection | Mobsuite | 3.1.9 | |
| 5. MLST detection | tseemann/MLST | 2.23.0 | |
| Visualization Module | |||
| 1. Circular genome view | Cgview | 2.0.3 | |
| 2. Assembly graph view | Bandage | 0.9.0 | |
| If SAM file, convert to BAM | SAMTOOLS | 1.21 | |
| 3. Read alignment coverage | tinycov | 0.4.0 | |
| 4. MLST distribution (pie chart) | Custom Python script | Python v3.12 | |
| 5. Heatmap of genes in common | Custom Python script | Biopython v1.81 | |
| 6. Plasmids and their lengths | Custom Python script | Pandas v2.0.0 | |
| 7. Number of reads and Average read quality | Custom Python script | Seaborn v0.12.0 | |
| 8. Binary heatmap of AMR/virulence genes | Custom Python scripts | Pandas v2.0.0 | |
| Database Download Module | |||
| 1. Bakta database | bakta_db | 1.10.4 | |
| 2. AMRFinderPlus database | amrfinder_update | 3.12.8 | |
| 3. CheckM2 database | CheckM2 | 14897628 | |
| 4. RGI database | RGI | 6.0.3 | |
| 5. Busco database | Busco | 5.8.3 (bacteria_odb10) | |
| 6. Kraken2 database | WGET | 8gb standard Kraken2 database | |
| 7. Gambit database | WGET | 1.0 |
BactiSeq: Documentation
BactiSeq documentation is split into the following pages:
- Getting started
- An overview on the pre-requisites, and how to install and use the pipeline
- BactiSeq Databases
- An overview on the type of databases necessary for the pipeline to run
- Usage
- An overview of how the pipeline works, how to run it and a description of all of the different command-line flags.
- Output
- An overview of the different results produced by the pipeline and how to interpret them.
You can find a lot more documentation about installing, configuring and running nf-core pipelines on the website: https://nf-co.re